 中文
中文Monoclonal antibody (mAb) drugs have developed rapidly in recent years, but their long R&D cycles and high costs limit patient access. mAb biosimilars have similar safety and efficacy profiles to the reference product. The development of a biosimilar follows a stepwise approach, including quality similarity, non‑clinical similarity, and clinical similarity studies. This stepwise process reduces uncertainty in clinical studies, increases the success rate of drug development, and improves patient access. Therefore, many biotech companies consider mAb biosimilars a key R&D focus. Quality attribute analysis of the reference product and similarity studies of the candidate drug are the foundation of biosimilar development and provide an important basis for clinical study design.
This article combines domestic and international regulatory requirements for biosimilars, biosimilar development experience, and global filing practices to comprehensively describe the content of quality similarity studies for mAb biosimilars. It particularly summarizes challenges in evaluating critical quality attributes, serving as an important reference for process development, helping companies accelerate drug development, and providing technical support to regulatory agencies.
Monoclonal antibody drugs originated in the 1980s and, after more than 30 years of development, have become the largest category of biologics. Among the top ten best‑selling drugs globally in 2019, six were mAbs. To enable more people to access high‑quality mAb therapies, scientists and pharmaceutical companies have pursued various ways to reduce costs. Among them, the development and approval of biosimilars will greatly reduce the patient burden and increase the accessibility of mAb drugs.
When developing and filing mAb biosimilars, many challenges arise, such as selection of the reference product (batch, quantity, shelf life, etc.), application of analytical methods, similarity evaluation principles, and assessment of product quality attributes. This article discusses these issues, aiming to provide ideas for mAb biosimilar development.
1. Quality Similarity Studies
Due to the complexity of mAb manufacturing processes and molecular structures, it is practically impossible to produce a product identical to the reference product. How to assess the similarity between a biosimilar and the reference product in terms of quality, safety, and efficacy is a problem that must be solved during biosimilar development. China ranks first globally in the number of biosimilars under development. Based on practical experience in mAb biosimilar development, the author summarizes the analysis of quality similarity into the following four aspects.
1.1 Basic Principles
According to the Guidelines for the Development and Evaluation of Biosimilars, the basic principles of biosimilar development and evaluation are: comparative principle, stepwise principle, consistency principle, and similarity evaluation principle. The Guidelines for Similarity Evaluation and Indication Extrapolation of Biosimilars issued in 2021 align with European and US guidelines, including the principles of overall similarity and stepwise progression.
Overall similarity focuses on a complete chain of evidence, including comprehensive evaluation of results from comparative studies on pharmaceutical quality, non‑clinical, and clinical aspects. The stepwise principle means that if differences exist between the candidate drug and the reference product in early studies, the impact of those differences on safety and efficacy should be assessed. If the impact is uncertain, targeted experiments should be designed in subsequent comparative studies.
1.2 Considerations for the Reference Product
The acceptance criteria for similarity evaluation of biosimilar quality attributes are determined based on the quality analysis results of the reference product. Several aspects should be considered when selecting a reference product.
1.2.1 Batch Requirements
The requirements for the reference product in the similarity guidelines of the European EMA and the US FDA are generally consistent. Taking the latest FDA guidance as an example, quality similarity studies should include at least 10 batches of the reference product, covering the shelf‑life range of the reference product. If fewer than 10 batches must be used for special reasons, early communication with the FDA is required for assessment. For the candidate biosimilar, 6–10 batches representative of commercial manufacturing processes should be included, typically covering batches used in key clinical studies and process validation. If process changes occur between batches of the candidate drug used for similarity studies, process comparability studies must be provided to ensure consistency in quality, safety, and efficacy across different process products.
1.2.2 Source Considerations
In early development, reference products from multiple sources are usually collected as references for process development and quality studies of the candidate drug. At the time of biosimilar marketing application, China’s NMPA, the European EMA, and the US FDA all require that the reference product used in quality similarity studies be sourced from the same region as the filing. If a reference product from a non‑filing region is used in similarity assessments, supporting data on the similarity between the candidate biosimilar and reference products from different sources must be provided.
The latest FDA guidance clearly states that all data and information for the reference product and biosimilar used in comparative studies (including physicochemical analysis, functional studies, animal studies, and clinical studies) should be included in the BLA dossier. If any batch is excluded from similarity studies, corresponding justification must be provided.
1.2.3 Reference Product Management
Beyond batch quantity and source, storage conditions and management procedures for the reference product also need attention. Based on practical experience, procurement of reference products in China currently takes a long time, and reference products may expire during head‑to‑head analysis experiments. To address this, samples can be frozen before expiry, provided there is a corresponding protocol and experimental evidence demonstrating that freezing does not affect product quality attributes – an approach accepted by domestic and international regulators. During on‑site inspections for marketing applications, reviewers also pay attention to reference product management, including procurement, aliquoting, storage, and usage records, to ensure the authenticity and traceability of reference product data in quality similarity studies.
1.3 Analytical Methods
Quality similarity studies are a major component of biosimilar registration filings and require high applicability and robustness of the analytical methods used:
① The methods must be “state‑of‑the‑art” – the most advanced in the current industry and technical field – with sufficient sensitivity to detect subtle differences in critical quality attributes between the biosimilar and reference product, especially the ability to identify degradation products in stability comparisons (i.e., “stability‑indicating” capability);
② For the same quality attribute, orthogonal analytical methods and techniques should be selected, such as methods based on different principles (physical, chemical, or biological);
③ In addition to routine release testing of drug substance (DS)/drug product (DP), quality similarity studies should include more comprehensive structural and characteristic characterization. Such analytical methods, due to their nature, do not require the same validation as release methods, but their rationality, reliability, and reproducibility should be scientifically demonstrated. Based on literature reports, regulatory review requirements, and practical experience, analytical methods commonly used for quality similarity are summarized in Table 1 (see original article for detailed table).
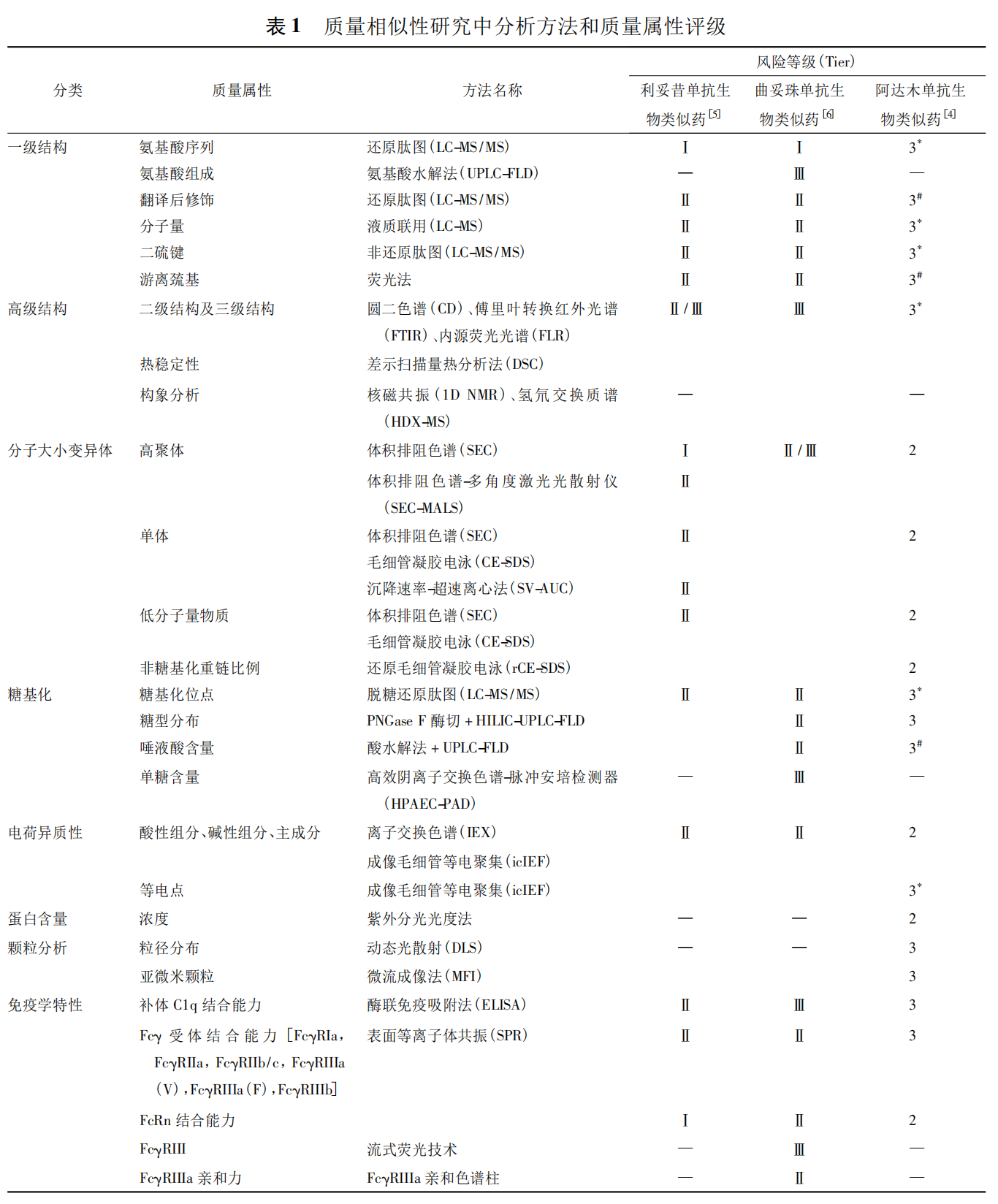
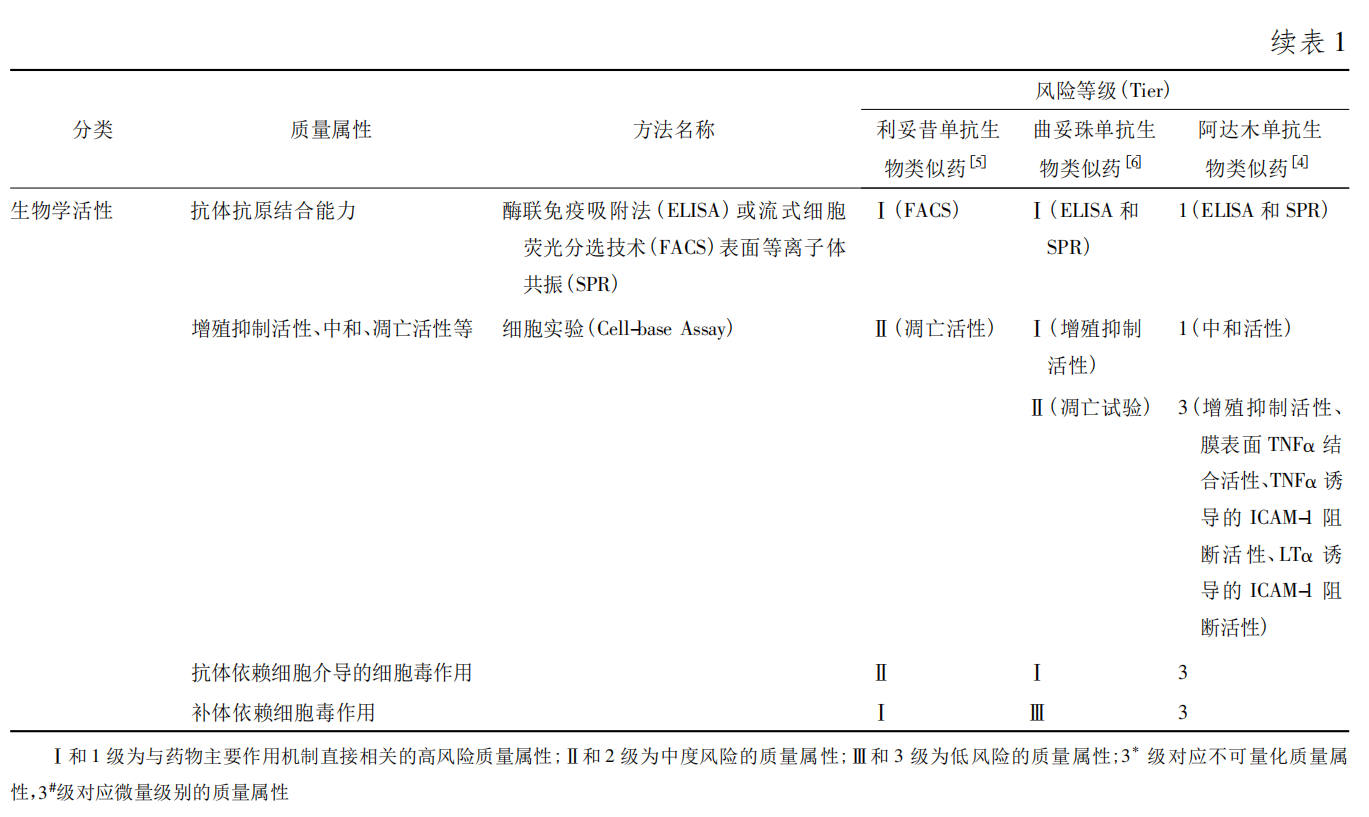
1.4 Similarity Acceptance Criteria for Quality Attributes
The US FDA issued a draft guidance in 2017 (since withdrawn) that described attribute tiering (Tier) for quality similarity assessment and corresponding assessment strategies, including statistical analysis methods to define similarity margins. In 2019, the FDA issued its latest guidance on quality similarity assessment for biosimilars, emphasizing the quality range approach to define similarity margins, with more flexible assessment methods. The EMA’s 2017 guidance introduced statistical assessment methods, including the Min‑Max method, Tolerance Intervals method, and quality range method (X‑sigma), consistent with the FDA’s description. Attribute tiering should be based on a comprehensive understanding of product quality and follow a “case‑by‑case” concept; the examples in Table 1 (original article) may serve as reference. It is important to note that while inferential statistical methods (e.g., equivalence analysis) are widely used in clinical studies, their application in quality attribute assessment may yield false positives due to sample size and selection effects.
When setting similarity evaluation criteria based on reference product quality data, factors such as changes in some quality attributes due to the reference product’s own process variations should be considered. Regulatory authorities have not given clear
recommendations on this phenomenon; company researchers must accumulate quality data trends for reference products at different stages and assess the impact of such changes on product safety and efficacy, as well as the reasonable setting of similarity criteria. In the quality similarity study of “Hanjuyou”, reference product batches with expiry dates from March 2017 to March 2021 showed changes in glycan ratios (especially galactosylation and afucosylation), and the corresponding ADCC and FcγRIIIa binding activities showed consistent changes. After evaluation, it was determined that these changes were related to process changes in the reference product and did not affect product safety or efficacy.
2. Key Considerations in Quality Similarity Studies
Quality similarity is the foundation of biosimilar development. The quality attributes of the reference product must be analyzed, and the biosimilar’s process developed accordingly. Key quality attributes to focus on in mAb quality similarity studies include primary structure, higher‑order structure, glycosylation, protein content, charge heterogeneity, molecular size variants, particle analysis, impurity analysis, biological activity, and immunological properties.
2.1 Primary Structure
Primary structure refers to the amino acid sequence and is the basis for higher‑order structure and biological function. In principle, the amino acid sequence of a biosimilar should be identical to that of the reference product. Quality attribute analyses related to primary structure typically include amino acid sequence, protein molecular weight, post‑translational modifications, disulfide bonds, and free sulfhydryl groups.
2.1.1 Amino Acid Sequence and Molecular Weight
Primary structure characterization methods are mostly based on LC‑MS, analyzing the molecule at different levels, supplemented by orthogonal methods. From the three dimensions of intact molecule, subunit level, and peptide level, the amino acid sequence consistency between the biosimilar and reference product can be comprehensively compared. Some methods, such as amino acid quantification and Edman degradation, can serve as orthogonal methods to demonstrate amino acid sequence consistency. In addition to amino acid sequence, it is also very important whether the inter‑chain and intra‑chain disulfide bond linkages of the biosimilar and reference product are consistent with theory. This is generally assessed by disulfide bond identification and quantitative determination of free sulfhydryl groups.
2.1.2 Disulfide Bonds
Disulfide bond analysis is generally performed by non‑reducing peptide mapping. Samples are sequentially digested with Lys‑C and trypsin to obtain disulfide‑linked peptides, which are analyzed by peptide molecular weight and tandem mass spectrometry, then compared with theoretical sequences to confirm consistency in disulfide bond linkage patterns between the biosimilar and reference product. Quantitation of free sulfhydryl groups is performed using Ellman’s reagent or other fluorescent dyes that react with sulfhydryl groups; the results also indirectly reflect consistency in disulfide bond structure.
2.1.3 Post‑translational Modifications
Post‑translational modifications are one cause of mAb heterogeneity and also affect biological activity and function. Consistency in the sites and levels of post‑translational modifications between the biosimilar and reference product is an important part of quality similarity studies. For post‑translational modifications other than glycosylation, they are categorized by site and modification type.
2.1.3.1 Deamidation
Deamidation of asparagine and glutamine to aspartic acid and glutamic acid, respectively, is a common post‑translational modification. In normal human IgG1 and IgG2, asparagine at positions 315 and 384 is prone to deamidation; it has been reported that about 23% of Asn384 undergoes deamidation. Deamidation of glutamine is common in the constant region of antibodies. Deamidation can occur at multiple sites in an antibody molecule, with varying sensitivity depending on the antibody’s higher‑order structure. If the antibody contains an NG motif, it is susceptible to sample handling and may produce a high proportion of post‑translational modification. Therefore, in quality similarity studies, attention should be paid to the sites and levels of deamidation and their impact on the safety and efficacy of the mAb.
2.1.3.2 Oxidation
Oxidation of methionine (M) in the complementarity‑determining region (CDR) may affect antigen‑binding activity; oxidation in the Fc region may affect thermal stability, Protein A binding, and FcRn binding activity. Therefore, when evaluating oxidation modifications, both the sites and levels of oxidation should be considered.
2.1.3.3 N‑terminal Modification
The N‑terminus of mAbs often undergoes cyclization of glutamine or glutamic acid to form pyroglutamic acid, a common modification in vivo. It is generally considered not to affect biological activity or function, but it is associated with charge heterogeneity. In similarity studies, it can be assessed in combination with charge heterogeneity analysis results.
2.1.3.4 C‑terminal Modification
The C‑terminus of the heavy chain of serum antibodies is usually glycine, but the gene encoding IgG shows a C‑terminal lysine. Studies have shown that an endogenous carboxypeptidase B in the body removes the C‑terminal lysine. Therefore, in quality similarity studies, it is generally accepted that removal of C‑terminal lysine does not affect product safety or efficacy. During mAb manufacturing, incomplete removal of C‑terminal lysine can lead to changes in charge heterogeneity. Thus, the impact of this modification should be considered in similarity assessment of charge heterogeneity. Other modifications such as isomerization, cysteinylation, and glycation should be assessed for similarity in combination with their levels, sites, and related activity and function studies.
2.2 Higher‑Order Structure
Higher‑order structure is crucial for the biological function of mAb drugs. Orthogonal techniques should be used to assess similarity from various aspects, including secondary and tertiary structure analysis.
Secondary structure is generally analyzed by far‑UV circular dichroism and infrared spectroscopy; tertiary structure is analyzed by near‑UV circular dichroism. Differential scanning calorimetry reflects structural changes through protein thermal stability; fluorescence spectroscopy reflects the spatial position and environment of specific amino acids (e.g., tryptophan); protein NMR depends on all higher‑order structural forms, including chemical shifts caused by amino acid type, secondary structure, tertiary structure, and kinetic differences in hydrogen‑deuterium exchange; ultracentrifugation reflects differences in higher‑order structure in solution by comparing sedimentation rates of monomers, fragments, and aggregates in their native state.
Since most higher‑order structure analytical methods are qualitative, the comparison is often based on spectral similarity; alternatively, empirical models can be used to further analyze spectra and obtain parameters for similarity comparison. Empirical models are mostly derived from literature or self‑built, so the source of the calculation software or model should be noted in the analysis results. For example, using Thermo OMINC to calculate spectral similarity, values closer to 1 indicate higher similarity. However, this method requires multiple experiments and is insensitive to small signal differences; visual comparison of spectra is still needed. Scientists at Amgen proposed a weighted spectra difference (WSD) calculation formula that reflects spectral differences more sensitively by calculating weighted deviations. For higher‑order structure analysis, the application of statistical analysis methods is likely to become a trend.
2.3 Glycosylation
Therapeutic monoclonal antibody products are glycoproteins. Glycosylation of mAbs typically occurs in the Fc region and, less commonly, in the Fab region. In similarity studies, glycosylation is evaluated primarily from the perspectives of efficacy and safety. The impact of glycosylation on efficacy is reflected in how different glycoforms affect antibody spatial structure, thereby influencing Fc‑mediated effector functions. The mechanism of action (MoA) varies among different mAbs, so similarity evaluation requirements for glycan species also differ accordingly. For IgG2/IgG4 subtype mAbs, the CH2 domain lacks amino acid sequences specific for Fcγ receptor and complement C1q binding, resulting in weak ADCC and CDC activities. Such antibodies are generally used to neutralize antigens or block receptor‑ligand binding. Therefore, the risk levels for afucosylated (Afuc) and galactosylated (Gal) glycans on these antibodies are low; in similarity evaluation, the main consideration is whether these attributes reflect structural integrity and process consistency. For IgG1 antibodies, ADCC and CDC are typical MoAs (e.g., rituximab). Similarity evaluation of Gal, Afuc, and high mannose (Man) should be given high priority; however, the case‑by‑case principle still applies. For example, trastuzumab and bevacizumab: the former has high HER2 expression that inhibits the complement pathway and lacks CDC, while the latter targets a soluble antigen and lacks both ADCC and CDC in its MoA. For these, minor differences in glycan patterns do not affect product efficacy. Some studies have also shown that antibodies with a high proportion of Man5 modification have reduced serum half‑life.
The safety impact of glycosylation is mainly immunogenicity, manifested as terminal sialic acid and α‑1,3‑linked galactose. Sialic acid exists in two forms: N‑glycolylneuraminic acid (NGNA) and N‑acetylneuraminic acid (NANA). NANA‑type sialic acid is commonly present in normal human serum antibodies, whereas anti‑NGNA antibodies exist in humans. Reusch et al. reported immune responses triggered by anti‑NGNA antibodies. Some antibodies expressed in murine cell lines may contain NGNA and α‑1,3‑linked galactose modifications. However, for most mAb products expressed in Chinese hamster ovary cells, the levels of NGNA and α‑1,3‑linked galactose are typically below the detection limit of analytical methods. In such cases, the rating for similarity studies may be considered medium.
2.4 Content
Content is an important aspect of similarity evaluation. In principle, the formulation and strength of a biosimilar should be the same as those of the reference product. Therefore, similarity in protein concentration should be evaluated. The EMA biosimilar guideline explicitly states that “strength” must be similar. For liquid or lyophilized formulations, protein concentration is typically determined by UV spectrophotometry. In head‑to‑head comparisons, the extinction coefficient is also very important.
The extinction coefficient of a protein is generally determined by its amino acid sequence. In early development, theoretical extinction coefficients are often used for concentration determination. Once the process reaches a certain stage (process parameters locked), the extinction coefficient should be experimentally confirmed. Multiple methods, such as amino acid quantification, Kjeldahl determination, and the Edelhoch method, are recommended for measuring and evaluating the extinction coefficient, establishing acceptance criteria, and finally determining the coefficient. For biosimilars, the extinction coefficient cannot be back‑calculated solely from the labeled concentration of the commercial product; it must be determined experimentally.
2.5 Charge Heterogeneity
During production, storage, and transportation, antibodies undergo post‑translational modifications as well as aggregate and fragment formation, leading to the generation of protein variants with different isoelectric points (pI). Similarity evaluation has high requirements for charge heterogeneity. First, chromatographic techniques (suitable for IgG1 antibodies) or free‑flow electrophoresis (suitable for IgG2 and IgG4 antibodies) should be used to prepare and enrich acidic and basic variants, identify their causes, establish structure‑function relationships (SFR), and evaluate the impact of each variant on safety and efficacy. Second, suitable analytical methods should be established to quantify, statistically calculate, and evaluate similarity of each acidic/basic component. Charge variant analysis of antibodies can be performed using ion exchange chromatography (IEX), based on differences in surface charge, or capillary isoelectric focusing (cIEF), based on differences in pI. Charge heterogeneity is greatly influenced by the protein expression platform (cell line and culture process) and purification process. Process development must balance production yield and similarity evaluation. For example, basic components caused by different degrees of C‑terminal lysine clipping (as described earlier) do not affect clinical safety or efficacy. In similarity evaluation, samples can be tested after CpB enzyme treatment.
2.6 Molecular Size Variants
Molecular size variants are product‑related impurities, mainly high molecular weight species (HMWS) and low molecular weight species (LMWS). They have potential safety implications and are quality attributes that must be strictly controlled. HMWS are identified using multi‑angle light scattering (MALS) and analytical ultracentrifugation (AUC); quantitative analysis typically employs two orthogonal techniques: size exclusion chromatography (SEC) and AUC. LMWS are assessed using capillary electrophoresis sodium dodecyl sulfate (CE‑SDS). In similarity assessment, it is first necessary to confirm that the types and levels of HMWS and LMWS in the candidate drug are similar to those in the reference product; otherwise, they must be identified and evaluated. Second, the quantitative analytical methods for purity and impurities must be validated, particularly the limit of quantitation (LOQ) or limit of detection (LOD). Finally, similarity assessment criteria should be calculated based on analytical data from at least 10 batches of the reference product. For impurity ranges, a one‑sided limit may be used, but should not exceed the release specification range.
2.7 Particle Analysis
Protein particles are an important quality attribute of antibody drugs and are closely related to product safety. Particles with sizes 100 nm – 1 μm are defined as submicron particles, 1–100 μm as sub‑visible particles, >100 μm as visible particles, and <100 nm as soluble aggregates or protein molecules. Typically, visible particles and aggregates are strictly monitored and controlled to very low levels during production. For sub‑visible particles, in vitro studies suggest they may cause immunogenicity and carry a risk of capillary blockage.
In quality similarity assessment, particle analysis is usually not performed; it needs to be monitored and evaluated during release and stability studies of the candidate drug. However, some domestic and international biosimilar similarity studies have included such comparisons. For example, Amgen used micro‑flow imaging (MFI) and light obscuration (LO) to compare sub‑visible particles of bevacizumab and adalimumab biosimilars, and used dynamic light scattering (DLS) and field flow fractionation (FFF) to compare submicron particles. Shanghai Henlius used DLS and MFI to analyze the particle profiles of trastuzumab and its biosimilar.
2.8 Impurity Analysis
According to ICH Q6B, impurities in mAb products can be divided into product‑related impurities and process‑related impurities. In similarity assessment, the evaluation of product‑related impurities focuses on their impact on safety and efficacy. Process‑related impurities, like product‑related impurities, can cause safety risks and must be strictly controlled. These include residual host cell proteins, host cell DNA, antibiotics, glycan modulators, protectants, etc., introduced during cell line construction and cell culture; residual Protein A introduced during downstream processing; and endotoxins and leachables present in the final drug product. The types and residual levels of process‑related impurities depend on the specific process. These impurities do not need to be similar to those of the reference product, but must be detected and appropriately controlled using advanced, sensitive analytical methods to ensure they are within safe limits. Therefore, process‑related impurities are not within the scope of similarity studies.
2.9 Biological Activity
Biological activity is related to the drug’s MoA. Biological activity analysis is a key focus of similarity evaluation. Based on antibody structure, it can be divided into Fab, Fc, and Fab/Fc‑mediated functions. Antigen‑binding activity is the foundation of mAb activity and belongs to the Fab functional region. Biological functions of the Fc region are introduced in the immunological properties section. Fab/Fc‑mediated biological functions include ADCC, CDC, and antibody‑dependent cellular phagocytosis (ADCP).
Biological activity assessment should be based on the drug’s MoA and follow the case‑by‑case principle for similarity evaluation. For example, IgG1 antibodies have strong Fcγ receptor and C1q binding ability, hence strong ADCC and CDC activity; for rituximab, the similarity rating is usually high or medium. For trastuzumab, ADCC is rated high in similarity evaluation, but cells with high HER2 expression inhibit the complement pathway, making CDC negative, so CDC is rated low. For adalimumab, the MoA is specific binding to soluble TNFα to block its interaction with cell surface TNFα receptors; therefore, neutralization activity is rated high, while ADCC and CDC are rated low. For antibodies that use Fab‑mediated specific binding to block endothelial/epidermal growth factor receptor pathway signaling (e.g., bevacizumab and trastuzumab), cell proliferation inhibition activity should be rated high. For IgG2 and IgG4 antibodies that have low Fc‑mediated effector cell killing, blocking activity should be rated high (e.g., anti‑PD‑1 antibodies). The Guidelines for Similarity Evaluation and Indication Extrapolation of Biosimilars also state that for indication extrapolation, appropriate biological activity comparison protocols should be designed according to the indication type.
Biological activity is closely related to quality attributes such as post‑translational modifications, glycosylation levels, and higher‑order structure. When there is uncertainty about similarity in these attributes, biological activity can be used for further explanation and supporting evidence.
2.10 Immunological Properties
Antibodies are key components of the adaptive immune system, and their relatively conserved Fc region has specific immunological properties. The Fc‑mediated biological functions of therapeutic antibodies include evaluation of Fc receptor binding ability and C1q binding ability.
FcRn participates in the recycling of serum IgG in vivo, affecting serum half‑life and consequently pharmacodynamics. In similarity studies, antibody FcRn binding ability is usually rated as medium.
C1q is the first binding protein in the classical complement activation pathway; binding to the Fc region of IgG initiates the CDC effect. FcγRIIIa binding ability is closely related to high mannose and afucosylation modifications, thereby affecting ADCC activity. C1q and FcγRIIIa binding abilities are typically defined as medium in similarity assessment. In specific cases, even if ADCC and CDC are not the MoA, C1q and FcγRIIIa binding abilities can assess Fc glycan structure and antibody higher‑order structural integrity, so they are also rated as medium. FcγRIa, FcγRIIa(R), FcγRIIa(H), FcγRIIb/c, and FcγRIIIb are less closely associated with glycan modifications than FcγRIIIa and are usually rated as low.
It is worth noting that antibody binding ability to FcγRIIIa is generally considered to correlate with Man% and Afuc% in glycan profiles, but a comprehensive assessment should consider the glycan ratios. For example, in Amgen’s bevacizumab biosimilar, although minor differences in Man% and Afuc% were observed between the biosimilar and reference product, the FcγRIIIa binding results showed high similarity. Therefore, minor differences in Man% and Afuc% do not affect product efficacy.
2.11 Stability Studies
Both domestic and international biosimilar guidelines require head‑to‑head stability studies between the biosimilar and reference product, including accelerated stability and forced degradation studies, to observe similarity in degradation trends and pathways.
According to the Technical Guidelines for Stability Studies of Biological Products (Trial), for products stored at 4°C, accelerated stability studies are typically conducted at 25°C. The number of batches, study duration, and test items for the selected biosimilar and reference product should be chosen based on the experimental design and objectives. Ultimately, similarity in stability trends under accelerated conditions can be observed between the biosimilar and reference product. Forced degradation studies include conditions such as high temperature, light, strong acid, strong base, shaking, oxidation, and freeze‑thaw. After exposure to these conditions, similarity comparisons are made by monitoring changes in primary structure, purity and impurities, charge heterogeneity, biological activity, etc. It is worth noting that regarding product shelf life, the EMA guideline recommends determining it based on the biosimilar’s own stability study results, without needing to compare shelf life with the reference product.
3. Conclusions and Outlook
Quality similarity is the prerequisite for biosimilar research and development. It determines the direction of process development and provides a reference for the design of non‑clinical and clinical study protocols. Based on years of experience in biosimilar development and global filings, and summarizing the latest domestic and international biosimilar regulations, this article presents a comprehensive overview of quality similarity studies for antibody biosimilars. It systematically introduces the stepwise and overall similarity principles, and provides a thorough description of quality similarity research and evaluation – covering reference product selection, analytical methods, similarity criteria, and key study considerations – serving as a reference for antibody R&D companies. The domestic and international biosimilar market is huge. Pharmaceutical companies’ technologies and platforms are maturing, and the regulatory system is improving. How to achieve standardized and systematic guidance – such as sharing reference product quality information, harmonizing similarity evaluation criteria, optimizing targets and capacity, creating data sharing and macro‑level capacity guidance, and avoiding resource waste – represents the new challenges and opportunities that pharmaceutical companies and regulatory agencies will face in the future.
Canton Biologics Biosimilar Case Study
As an internationally leading biologics CDMO, Canton Biologics has unique technical advantages in biosimilar development. Developing a biosimilar requires establishing quality standards based on the reference product to achieve quality comparability; it also requires navigating around the reference product’s patent protections and re‑establishing analytical methods to meet filing requirements.
Highlights:
-
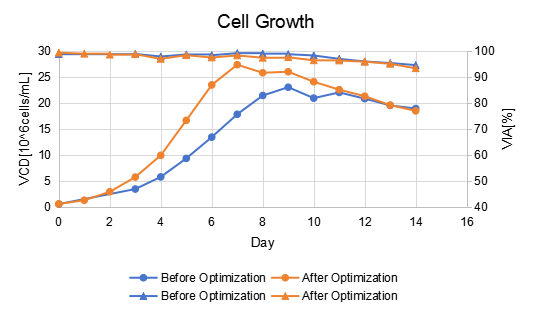
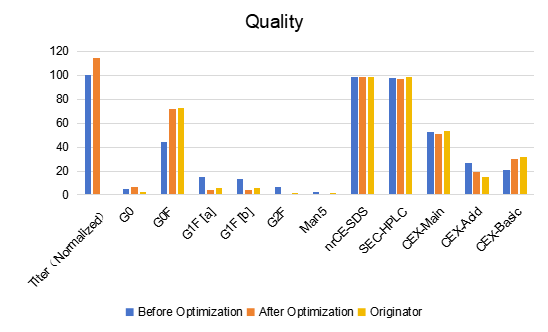
In one project, after process optimization by Canton Biologics, a biosimilar achieved comparability to the reference product with an expression titer of 6–7 g/L (Figure 1).
-
This project has already obtained an IND filing approval for the biosimilar.
-
Through culture additives, feeding strategy optimization, and process optimization, the protein quality was adjusted to be consistent with the reference product.